 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeLatent Spherical Flow Policy for Reinforcement Learning with Combinatorial Actions
Jan 29, 2026Reinforcement learning (RL) with combinatorial action spaces remains challenging because feasible action sets are exponentially large and governed by complex feasibility constraints, making direct policy parameterization impractical. Existing approaches embed task-specific value functions into constrained optimization programs or learn deterministic structured policies, sacrificing generality and policy expressiveness. We propose a solver-induced \emph{latent spherical flow policy} that brings the expressiveness of modern generative policies to combinatorial RL while guaranteeing feasibility by design. Our method, LSFlow, learns a \emph{stochastic} policy in a compact continuous latent space via spherical flow matching, and delegates feasibility to a combinatorial optimization solver that maps each latent sample to a valid structured action. To improve efficiency, we train the value network directly in the latent space, avoiding repeated solver calls during policy optimization. To address the piecewise-constant and discontinuous value landscape induced by solver-based action selection, we introduce a smoothed Bellman operator that yields stable, well-defined learning targets. Empirically, our approach outperforms state-of-the-art baselines by an average of 20.6\% across a range of challenging combinatorial RL tasks.
Learning simple heuristic rules for classifying materials based on chemical composition
May 05, 2025In the past decade, there has been a significant interest in the use of machine learning approaches in materials science research. Conventional deep learning approaches that rely on complex, nonlinear models have become increasingly important in computational materials science due to their high predictive accuracy. In contrast to these approaches, we have shown in a recent work that a remarkably simple learned heuristic rule -- based on the concept of topogivity -- can classify whether a material is topological using only its chemical composition. In this paper, we go beyond the topology classification scenario by also studying the use of machine learning to develop simple heuristic rules for classifying whether a material is a metal based on chemical composition. Moreover, we present a framework for incorporating chemistry-informed inductive bias based on the structure of the periodic table. For both the topology classification and the metallicity classification tasks, we empirically characterize the performance of simple heuristic rules fit with and without chemistry-informed inductive bias across a wide range of training set sizes. We find evidence that incorporating chemistry-informed inductive bias can reduce the amount of training data required to reach a given level of test accuracy.
Practical Efficiency of Muon for Pretraining
May 04, 2025We demonstrate that Muon, the simplest instantiation of a second-order optimizer, explicitly expands the Pareto frontier over AdamW on the compute-time tradeoff. We find that Muon is more effective than AdamW in retaining data efficiency at large batch sizes, far beyond the so-called critical batch size, while remaining computationally efficient, thus enabling more economical training. We study the combination of Muon and the maximal update parameterization (muP) for efficient hyperparameter transfer and present a simple telescoping algorithm that accounts for all sources of error in muP while introducing only a modest overhead in resources. We validate our findings through extensive experiments with model sizes up to four billion parameters and ablations on the data distribution and architecture.
Rethinking Reflection in Pre-Training
Apr 05, 2025



A language model's ability to reflect on its own reasoning provides a key advantage for solving complex problems. While most recent research has focused on how this ability develops during reinforcement learning, we show that it actually begins to emerge much earlier - during the model's pre-training. To study this, we introduce deliberate errors into chains-of-thought and test whether the model can still arrive at the correct answer by recognizing and correcting these mistakes. By tracking performance across different stages of pre-training, we observe that this self-correcting ability appears early and improves steadily over time. For instance, an OLMo2-7B model pre-trained on 4 trillion tokens displays self-correction on our six self-reflection tasks.
Predicting band gap from chemical composition: A simple learned model for a material property with atypical statistics
Jan 06, 2025



In solid-state materials science, substantial efforts have been devoted to the calculation and modeling of the electronic band gap. While a wide range of ab initio methods and machine learning algorithms have been created that can predict this quantity, the development of new computational approaches for studying the band gap remains an active area of research. Here we introduce a simple machine learning model for predicting the band gap using only the chemical composition of the crystalline material. To motivate the form of the model, we first analyze the empirical distribution of the band gap, which sheds new light on its atypical statistics. Specifically, our analysis enables us to frame band gap prediction as a task of modeling a mixed random variable, and we design our model accordingly. Our model formulation incorporates thematic ideas from chemical heuristic models for other material properties in a manner that is suited towards the band gap modeling task. The model has exactly one parameter corresponding to each element, which is fit using data. To predict the band gap for a given material, the model computes a weighted average of the parameters associated with its constituent elements and then takes the maximum of this quantity and zero. The model provides heuristic chemical interpretability by intuitively capturing the associations between the band gap and individual chemical elements.
Multimodal Learning for Crystalline Materials
Nov 30, 2023



Artificial intelligence (AI) has revolutionized the field of materials science by improving the prediction of properties and accelerating the discovery of novel materials. In recent years, publicly available material data repositories containing data for various material properties have grown rapidly. In this work, we introduce Multimodal Learning for Crystalline Materials (MLCM), a new method for training a foundation model for crystalline materials via multimodal alignment, where high-dimensional material properties (i.e. modalities) are connected in a shared latent space to produce highly useful material representations. We show the utility of MLCM on multiple axes: (i) MLCM achieves state-of-the-art performance for material property prediction on the challenging Materials Project database; (ii) MLCM enables a novel, highly accurate method for inverse design, allowing one to screen for stable material with desired properties; and (iii) MLCM allows the extraction of interpretable emergent features that may provide insight to material scientists. Further, we explore several novel methods for aligning an arbitrary number of modalities, improving upon prior art in multimodal learning that focuses on bimodal alignment. Our work brings innovations from the ongoing AI revolution into the domain of materials science and identifies materials as a testbed for the next generation of AI.
Topogivity: A Machine-Learned Chemical Rule for Discovering Topological Materials
Mar 02, 2022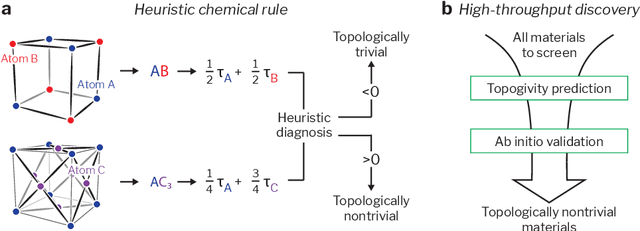
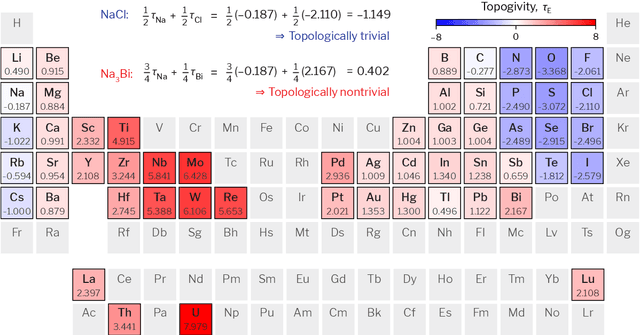
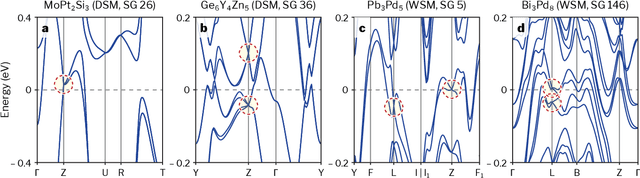
Topological materials present unconventional electronic properties that make them attractive for both basic science and next-generation technological applications. The majority of currently-known topological materials have been discovered using methods that involve symmetry-based analysis of the quantum wavefunction. Here we use machine learning to develop a simple-to-use heuristic chemical rule that diagnoses with a high accuracy whether a material is topological using only its chemical formula. This heuristic rule is based on a notion that we term topogivity, a machine-learned numerical value for each element that loosely captures its tendency to form topological materials. We next implement a high-throughput strategy for discovering topological materials based on the heuristic topogivity-rule prediction followed by ab initio validation. This way, we discover new topological materials that are not diagnosable using symmetry indicators, including several that may be promising for experimental observation.